Allergie allgemein
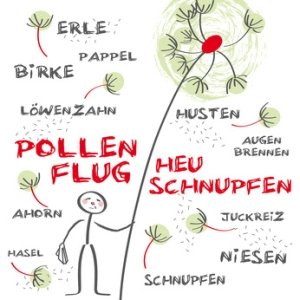
Die Allergiewelle rollt schon seit Jahren, Mediziner sprechen bereits von der “Volkskrankheit Allergie”. Dabei ist wichtig zu wissen: Eine Allergie kommt nicht immer allein. Die sogenannten “atopischen Erkrankungen” treten oft gemeinsam auf. Wissenswertes zu Ursachen, Symptomen, Diagnose und Therapie von Allergien ist unter “Allergie: Was ist das?” zusammengefasst.
Aktuelle Beiträge und Interviews mit Medizinern zu allgemeinen Allergiethemen finden Sie hier:




















- 1
- 2
News - Allergie Allgemein
- Update Allergologie - Immunologie: Was ist neu?
- 20 Jahre Allergologie im Kloster: Neues zu Diagnostik & Therapie
- Deutscher Allergiekongress 2023: Ein Blick in die Zukunft
- CFS: Wie kommt es zum chronischen Fatigue-Syndrom?
- Alternative Heilmethoden bei Allergien, geht das?
- Allergie – was ist das?
- Allergien dank molekularer Allergologie verhindern?
- Biologika bei Asthma, Neurodermitis, Urtikaria, Nasenpolypen
")
")
")
")

")
")
")
")
")

")
